Cecilia Lanzi, Chiara Aieta, Michele Ceotto, Riccardo Conte
J. Chem. Phys. 163, 024122 (2025)
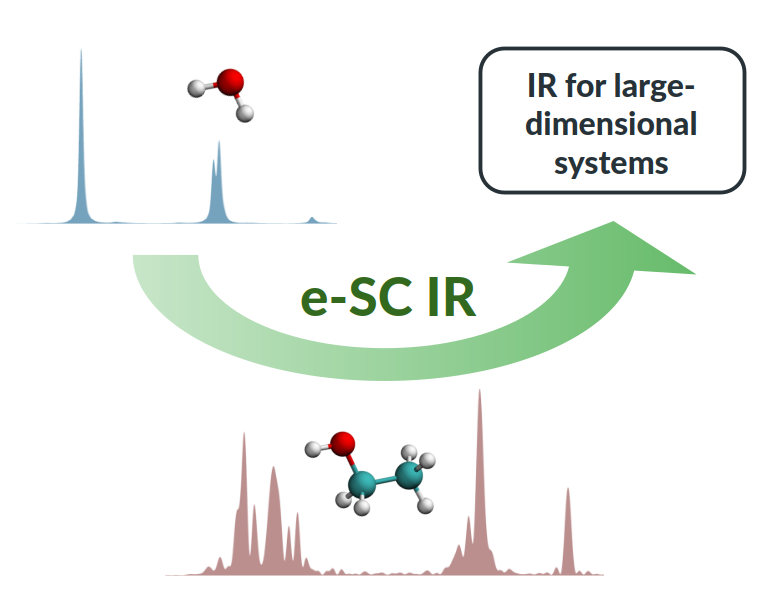
Recently—Lanzi et al., J. Chem. Phys. 160, 214107 (2024)—we introduced a time averaged approach to infrared (IR) spectroscopy. The pivotal advance in that paper was represented by the possibility to get accurate semiclassical estimates of the IR absorption intensities and associated transition frequencies from a single calculation. However, the method relies on the convergence of Monte Carlo integrations based on the generation of thousands of pairs of semiclassical trajectories. This makes the approach highly accurate but limited to small, few-atom molecules. Here, we build on the theoretical grounds of that work to extend the application of the method to larger molecules. The goal is achieved by moving to tailored single-pair trajectory calculations and introducing a partially time-independent approximation to the real part of the coherent state overlap. Upon testing the level of accuracy on small molecules such as water, formaldehyde, and methane, we calculate IR spectra for ethanol and glycine. Vibrational intensities and frequencies are found to be fairly accurate, and the method can be straightforwardly applied to larger molecular systems.